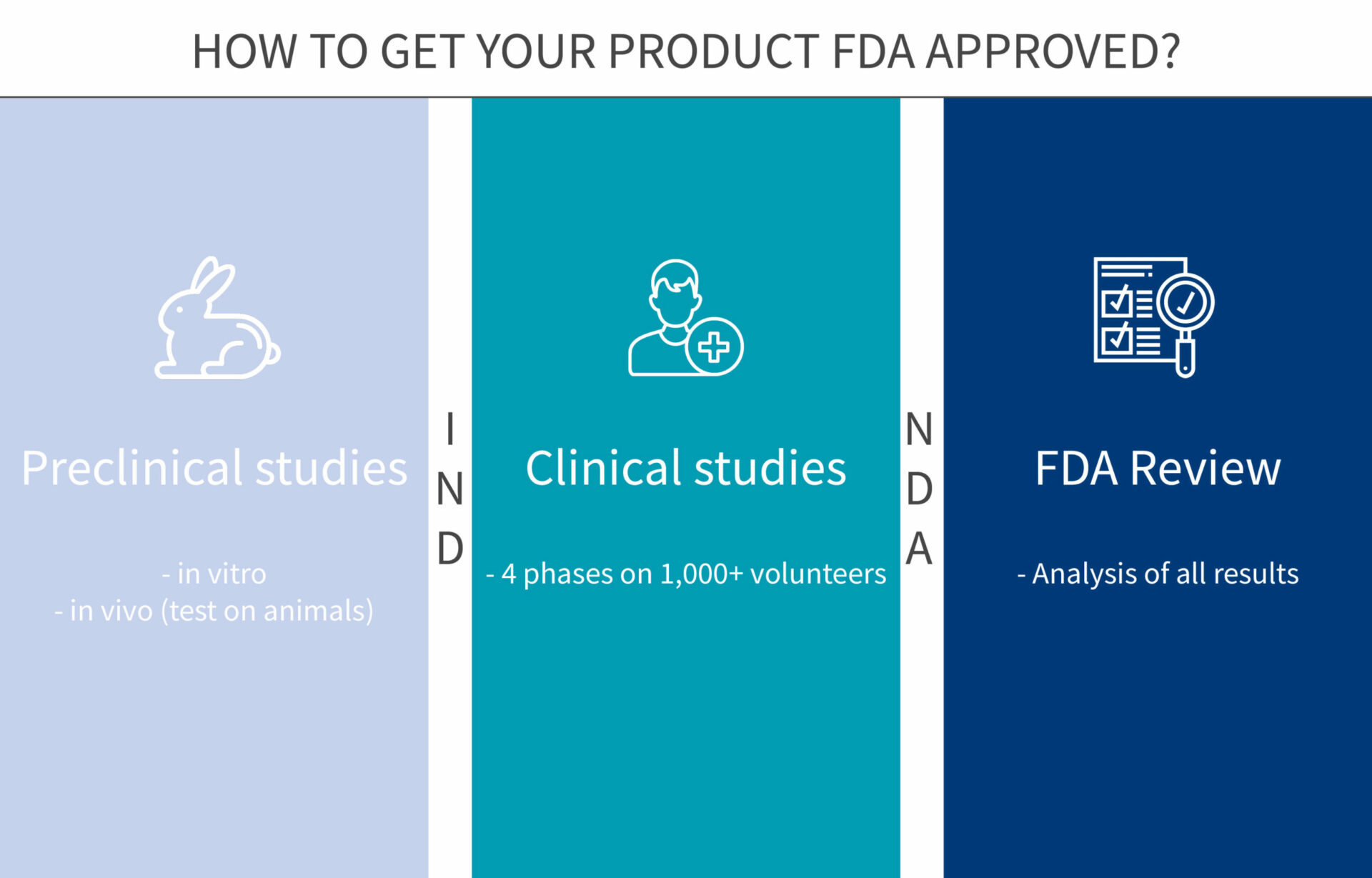
6 steps to get your new product approved by FDA
In the USA, any new drug or biologic needs a stamp of approval from the Food and Drug Administration (FDA) before going to the market. The development of a new drug and the approval process can be long and difficult. To make it easier for you, we have gathered below all the tips and information you will need to get through the 6 steps of the approval process.

Step 1: In vitro studies, gather info on the new compound
Aims:
- Provide results that will support an Investigational New Drug (IND) application
- Minimize the risks before clinical trials
Outcomes:
- First keys to understanding the new drug mechanism of action
- Preliminary toxicity profile
- Preliminary efficacy assessment
What does the FDA require?
In vitro results on 2D and 3D cell cultures are required in the IND application. However, it doesn’t take into account the cell environment and how the compound could interact with it. That’s why biotechs are developing more accurate in vitro assays. Testing on bio-printed tissue, organ-on-a-chip assays or organoids will most likely become the in vitro norm for the FDA.
Bio-printed tissue is one of the most popular new in vitro methods. The tissue is reconstructed thanks to a 3D printer filled with bio-ink, a gel carrying human-derived cells. This method can reconstruct the 3-dimensional structure of human tissue from most organs. Testing new drugs on these reconstructed tissues can provide a lot more information than 2D cells and more accurate results than animal testing. This technique is also able to model a patient’s tumor from actual tumor cells. This can be key to develop personalized cancer therapy.
3D bioprinted tissues. Credit: Paige Derr and Kristy Derr, National Center for Advancing Translational Science
Following similar ideas, drugs can now be tested on organoids, which are miniature organ models grown from stem cells. Organ-on-a-chip assays mimic the structure and behavior of real organs for similar testing.
All of these new in vitro techniques are still at an early stage of development, but they show promising results to support drug development.
Step 2: In vivo studies, get a comprehensive understanding of the drug
Aims:
- Gather information on the new compound
- Provide results that will support an Investigational New Drug (IND) application
- Minimize the risks before clinical trials
Outcomes:
- A comprehensive understanding of the new drug mechanism of action
- Complete toxicity profile
- Determination of no-observed-adverse-effect (NOAE) levels i.e. determination of a safe starting dose for clinical trials
- Efficacy assessment
What does the FDA require?
In vivo results on two mammalian species (usually one rodent and one non-rodent) are required for an IND application.
Requirements for in vivo preclinical studies include:
- Short term animal studies (acute) to determine the pharmacological action and toxicity of the new drug
- Long term animal studies (chronic) to look for potential side effects that may result from long-term use such as carcinogenicity or reproductive effects.
For these studies, the choice of animal is crucial as it will affect toxicology and efficacy results. For example, testing subcutaneously delivered drugs on mice will not yield reliable results as their skin is drastically different from humans.
History shows that compounds with promising results in animals can reveal themselves not as effective in humans and vice versa. Adverse effects observed in animals can also differ when the drug is translated into human trials. These interspecies differences lead to most clinical studies failures. Besides, experimenting on animals presents ethical and cost challenges. Consequently, regulatory agencies around the world tend to encourage the development of alternatives to animal studies.
What is recommended to minimize clinical study failure?
-
Ex vivo studies
Innovative ex vivo human assays are known as reliable alternatives to animal testing. They are already approved and widely used to replace animal testing in the cosmetic industry. Ex vivo studies consist of tests on real live human biopsies kept alive with appropriate culture conditions. Tests on human skin biopsies help determine local toxicity and efficacy of compounds. They are key in understanding the interactions occuring between the new drug and skin. Almost any kind of formulation can be tested on such assays as they can undergo topical application, systemic administration, and even subcutaneous and intradermal injections. Assay culture can last up to 10 days, which is long enough to determine toxicity and efficacy.
However, assays made from real human tissues are difficult to implement on organs other than skin. The challenge posed by the lack of blood circulation can be a limitation for the study of drug metabolization.

Genoskin’s ex vivo human skin assay, HypoSkin®
-
In silico studies
Computational modeling of structure-activity relationships could be key to help understand the new drug’s mechanism of action. Also, thanks to all the data already gathered by researchers, interactions between a compound and specific receptors can be modeled . These new computational models will be especially helpful to monitor any adverse effect before going into in vivo testing.
When the characterization of biotech alternatives demonstrates that they can be as predictive as animal testing, they would be a cheaper, safer and faster way to get a new drug on the market. This will happen. Regulatory agencies around the world are preparing for it. However, this may not completely end trials on animals. A final safety evaluation may probably still be required by the FDA before moving to trials in humans.

Step 3: Phase 1 of clinical trials, safety assessment in humans
Volunteers: 20 to 100 healthy volunteers (or volunteers with the condition for cancer treatment)
Duration: Several months
Aims:
- Determine what are the drug’s most frequent and serious adverse events
- Understand how the drug is metabolized and excreted
- Safety assessment and optimal dosage
Step 4: Phase 2 of clinical trials, efficacy assessment in humans
Volunteers: 300+ volunteers with the condition
Duration: Several months to 2 years
Aims:
- Obtain preliminary data on effectiveness
- Perform a comparison of the new drug with a placebo or another known drug
- Study safety and short term adverse effects
Step 5: Phase 3 of clinical trials, monitoring adverse reactions
Volunteers: 300 to 3,000 volunteers with the condition
Duration: 1 to 4 years
Aims:
- Gather even more information about safety and effectiveness
- Test the new drug on different populations with different dosages
- Test the new drug in combination with other drugs
- Monitor longer-term adverse reactions
Step 6: FDA Review
If proven safe and efficient through the first three phases of clinical trials, the new drug can be considered for an NDA (New Drug Application).
To pretend to FDA approval, the NDA must include:
- all information from preclinical data to phase 3 of clinical trials
- reports on all studies, data, and analyses performed inside or outside the USA
- a proposed labeling
- safety updates
- patent information
- institutional review board compliance information
- directions for use
Based on the NDA file, the FDA review team takes 6 to 10 months to review all the information. The review team also conducts a routine inspection of the clinical study site.
At the end of the review phase, the FDA gives some recommendations and may ask for additional information or studies. Once all the shadow areas are cleared, a senior FDA official makes the final decision. If the new drug is approved, it will then be allowed to be marketed.
A drug being marketed doesn’t mean the end of safety and efficacy monitoring. There is a fourth phase of clinical trials involving 3000+ volunteers for several years. This study will monitor the drug’s safety in the long term and potentially provide updates on the drug’s optimal usage.
To keep up-to-date with Genoskin’s latest news, follow us on Twitter and LinkedIn. You can also contact us to learn more about our products and services.
Comments are closed.



